
Quick Start with the wien2k code
Calculations of Solid Properties Using Wien2k Package :: WIEN2K ENVIRONMENT :: WIEN2K DOCUMENTATIONS
Page 1 of 1

Quick Start with the wien2k code
![]() by Algerien1970 Fri 22 May - 18:33
by Algerien1970 Fri 22 May - 18:33
This page is a summary of the official userguide of wien2k in this link : http://www.wien2k.at/features/3Quick_Start.html#fig:eplot
There are three main stages of calculation to do before the calculations of the different properties of a material. These stages are :
A/ First stage : Prepation of the Struct File
1/ Creating a new session :
The user interface w2web uses sessions to distinguish between different working environments and to quickly change between different calculations. First you have to create a new session (or select an old one). Enter ``TiC'' and click the ``Create'' button.
Note: Creating a session does not automatically create a new directory!
You will be placed in your home directory if no working directory was designated to this session previously (or if the directory does not exist any more).

2/ Creating a new case-directory
Using ``Session Mgmt. $\rightarrow $ change directory'' you can select an existing directory or create a new one. For this example create a new directory lapw and than TiC using the ``Create'' button. After the directory has been created, you have to click on select current directory to assign this newly created directory to the current session.
After clicking on Click to restart session the main window of w2web will appear (Fig.3.3.)

3/ Creating the ``master input`` file case.struct
To create the file TiC.struct start the struct-file generator using ``Execution $\rightarrow $ StructGen'' (see figure 3.4).
For a new case w2web creates an empty structure template in which you can specify structural data. Later on this information is used to generate the TiC.struct file.
As a first step specify the number of atoms (2 for TiC) and fill in the data given below into the corresponding fields (white boxes):
Click ``Save Structure'' (Z will be updated automatically) and ``set automatically RMT and continue editing '':
This will compute the nearest neigbor distances using the program nn and setrmt_lapw will then determine the optimal RMT values (muffin-tin radius, atomic sphere radius). To learn more about the philosophy of setting RMTs see $http://www.wien2k.at/reg_user/faq$. Since it is essential to keep RMTs constant within a series of calculations (eg. when you do a Volume-optimization, see 3.11.6 ), you should already now decide whether you want to do just one single calculation with fixed structural parameters, or whether you intend a relaxation of internal parameters (using forces and min_lapw) or a volume optimization, which would required reduced RMT values.
Choose a reduction of 3 % so that we can later optimize the lattice parameter.

When you are done, exit the StructGen with ``save file and clean up''. This will generate the file TiC.struct (shown now in view-only mode with a different background color), which is the master input file for all subsequent programs. This step also automatically generates the input file for the free atom program lstart (atomic configurations) tic.inst.
A few other hints on StructGen:
You have to click on Save Structure after every modifications you make in the white fields. Add/remove a position/atom only if you have made no other changes before.
In a face-centered (body-centered) spacegroup you have to enter just one atom (not the ones in (.5,.5,0),...).
StructGen offers a built in calculator: Each position of equivalent atoms can be entered as a number, a fraction (e.g. $1/3$) or a simple expression(e.g. $0.21+1/3$). The first position defines the variables x, y and z, which can be using in expression defining the other positions (e.g. $-y$, $x$, $-z+1/2$).
When you now choose ``Files $\rightarrow $ show all files'', you will see, that both files tic.struct and tic.inst have been created.
For a detailed description of these files consult sections 4.3 and 6.4.3.
There are three main stages of calculation to do before the calculations of the different properties of a material. These stages are :
A/ First stage : Prepation of the Struct File
1/ Creating a new session :
The user interface w2web uses sessions to distinguish between different working environments and to quickly change between different calculations. First you have to create a new session (or select an old one). Enter ``TiC'' and click the ``Create'' button.
Note: Creating a session does not automatically create a new directory!
You will be placed in your home directory if no working directory was designated to this session previously (or if the directory does not exist any more).
2/ Creating a new case-directory
Using ``Session Mgmt. $\rightarrow $ change directory'' you can select an existing directory or create a new one. For this example create a new directory lapw and than TiC using the ``Create'' button. After the directory has been created, you have to click on select current directory to assign this newly created directory to the current session.
After clicking on Click to restart session the main window of w2web will appear (Fig.3.3.)
3/ Creating the ``master input`` file case.struct
To create the file TiC.struct start the struct-file generator using ``Execution $\rightarrow $ StructGen'' (see figure 3.4).
For a new case w2web creates an empty structure template in which you can specify structural data. Later on this information is used to generate the TiC.struct file.
As a first step specify the number of atoms (2 for TiC) and fill in the data given below into the corresponding fields (white boxes):
Title TiC
Lattice F (for face centered)
a 4.328 Å(make sure the Ang button is selected)
b 4.328 Å
c 4.328 Å
$\alpha,\beta,\gamma$ 90
Atom Ti, enter position (0,0,0)
Atom C, enter position (.5,.5,.5)
Lattice F (for face centered)
a 4.328 Å(make sure the Ang button is selected)
b 4.328 Å
c 4.328 Å
$\alpha,\beta,\gamma$ 90
Atom Ti, enter position (0,0,0)
Atom C, enter position (.5,.5,.5)
Click ``Save Structure'' (Z will be updated automatically) and ``set automatically RMT and continue editing '':
This will compute the nearest neigbor distances using the program nn and setrmt_lapw will then determine the optimal RMT values (muffin-tin radius, atomic sphere radius). To learn more about the philosophy of setting RMTs see $http://www.wien2k.at/reg_user/faq$. Since it is essential to keep RMTs constant within a series of calculations (eg. when you do a Volume-optimization, see 3.11.6 ), you should already now decide whether you want to do just one single calculation with fixed structural parameters, or whether you intend a relaxation of internal parameters (using forces and min_lapw) or a volume optimization, which would required reduced RMT values.
Choose a reduction of 3 % so that we can later optimize the lattice parameter.
Figure: StructGen of w2web
When you are done, exit the StructGen with ``save file and clean up''. This will generate the file TiC.struct (shown now in view-only mode with a different background color), which is the master input file for all subsequent programs. This step also automatically generates the input file for the free atom program lstart (atomic configurations) tic.inst.
A few other hints on StructGen:
You have to click on Save Structure after every modifications you make in the white fields. Add/remove a position/atom only if you have made no other changes before.
In a face-centered (body-centered) spacegroup you have to enter just one atom (not the ones in (.5,.5,0),...).
StructGen offers a built in calculator: Each position of equivalent atoms can be entered as a number, a fraction (e.g. $1/3$) or a simple expression(e.g. $0.21+1/3$). The first position defines the variables x, y and z, which can be using in expression defining the other positions (e.g. $-y$, $x$, $-z+1/2$).
When you now choose ``Files $\rightarrow $ show all files'', you will see, that both files tic.struct and tic.inst have been created.
For a detailed description of these files consult sections 4.3 and 6.4.3.

Algerien1970- Forum Manager

- Messages : 485
Date d'inscription : 2015-05-14 -

Re: Quick Start with the wien2k code
![]() by Algerien1970 Fri 22 May - 18:42
by Algerien1970 Fri 22 May - 18:42
B/ Second stage : Initialization ( Preparation of the imput files )
After the two basic input files have been created, initalization of the calculation is done by ``Execution $\rightarrow $ initialize calc.''. This will guide you through the steps necessary to initialite the calculation. Simply follow the steps that are highlighted in green and follow the instructions.
The initialization process is described in detail in section 5.1.2.
Alternatively you could run the script init_lapw from the command line. All actions of this script are logged in short in :log and in detail in the file case.dayfile, which can easily be accessed by Utils. $\rightarrow $ show dayfile.
Initializing the calculation will run several steps automatically, where x is the script to start WIEN2k programs (see section: 5.1.1).
x nn
calculates the nearest neighbors up to a specified distance and thus helps to determine the atomic sphere radii (you must specify a distance factor f, e.g. 2, and all distances up to f * NN-dist. are calculated)
view TiC.outputnn
: check for overlapping spheres, coordination numbers and nearest neighbor distances, (e.g. in the sodium chloride structure there must 6 nearest and 12 next nearest neighbors). Using these distances and coordinations you can check whether you put the proper positions into your struct file or if you made a mistake. nn also checks whether your equivalent atoms are really crystallographically equivalent and eventually writes a new struct-file which you may or may not accept. If you have not done so at the very beginning, go back to StructGen and choose proper RMT values. You can save a lot of CPU-time by changing RMT to almost touching spheres. See Sec.4.3
x sgroup
calculates the point and spacegroups for the given structure
view TiC.outputsgroup
: Now you can either accept the TiC.struct file generated by sgroup (if you want to use the spacegroup information or a different cell has been found by sgroup) or keep your original file (default).
x symmetry
generates from a raw case.struct file the space group symmetry operations, determines the point group of the individual atomic sites, generates the LM expansion for the lattice harmonics (in case.in2_st) and local rotation matrices (in case.struct_st).
view TiC.outputs
: check the symmetry operations (they have been written to or compared with already available ones in TiC.struct by the program symmetry) and the point group symmetry of the atoms (You may compare them with the ``International Tables for X-Ray Crystallography``). If the output does not match your expectations from the ``Tables'', you might have made an error in specifying the positions. The TiC.struct file will be updated with symmetry operations, positive or negativ atomic counter (for ``cubic'' point group symmetries) and the local rotation matrix.
x lstart
generates atomic densities (see section 6.4) and determines how the orbitals are treated in the band structure calculations (i.e. as core or band states, with or without local orbitals, ...). You are requested to specify the desired exchange correlation potential and an energy that separates valence from core states. For TiC select the recommended potential option ``GGA of Perdew-Burke-Ernzerhof 96'' and a separation energy of -6.0 Ry.
edit TiC.outputst
: check the output (did you specify a proper atomic configuration, did lstart converge, are the core electrons confined to the atomic sphere?). Warnings for the radial mesh can usually be neglected since it affects only the atomic total energy. lstart generates TiC.in0_st, in1_st, in2_st, inc_st and inm. For Ti it selects automatically $1s$, $2s$, and $2p$ as core states, $3s$ and $3p$ will be treated with local orbitals together with $3d$, $4s$ and $4p$ valence states.
edit TiC.in1_st
: As mentioned, the input files are generated automatically with some default values which should be a reasonable choice for most cases. Nevertheless we highly recommend that you go through these inputs and become familiar with them. The most important parameter here is RKMAX, which determines the number of basis functions (size of the matrices). Values between 5-9 (APW) and 6-10 (LAPW) are usually reasonable. You may change here the usage of APW or LAPW (set 1 or 0 after the CONT/STOP switch), since often APW is necessary only for orbitals more difficult to converge (3d, 4f). Here we will just change EMAX of the energy window from 1.5 to 2.0 Ry in order to be able to calculate the unoccupied DOS to higher energies.
edit TiC.in2_st
: Here you may limit the LM expansion (for some speedup), change the value of GMAX (in cases with small spheres (e.g. systems with H-atoms) values of 15-24 are recommended) or specify a different BZ-integration method to determine the Fermi energy. For this example you should not change anything so that you can compare your results with the test run.
edit TiC.inm_st
: For ``difficult to converge systems'' (several atoms with localized d- or f-electrons, magnetic systems) you should reduce the mixing factor from 0.4 to a smaller value (e.g. 0.05). (See our faq-page on www.wien2k.at what you should do when the scf cycle crashes). For TiC no changes are necessary.
Copy all generated inputs
(from case.in$*$_st to case.in*). In cases without inversion symmetry the files case.in1c, in2c are produced.
x kgen
generates a k-mesh in the Brillouin zone (BZ). You must specify the number of k-points in the whole BZ (use 1000 for comparison with the provided output, a ``good'' calculations needs many more). For details see section 6.5.
view TiC.klist
: check the number of k-points in the irreducible wedge of the BZ (IBZ) and the energy interval specified for the first k-point. You can now either rerun kgen (and generate a different k-mesh) or continue.
x dstart
generates a starting density for the SCF cycle by superposition of atomic densities generated in lstart. For details see section 6.6.
view TiC.outputd
(check if gmax $>$gmin)
Now you are asked
, whether or not you want to run a spin-polarized calculation (in such a case case dstart is re-run to generate spin-densities). For TiC say No.
Alternatively, w2web provides an ``expert-mode'', where some inputs can be specified right at the beginning and then the whole initialization runs at once. Please check carefully the STDOUT-listing and some output-files for possible errors or warnings!!
Initialization of a calculation (running init_lapw) will create all inputs for the subsequent SCF calculation choosing some default options and values. You can find a list of input files using ``Files $\rightarrow $ input files'' ( 3.5).

Figure 3.5: List of input files
Algerien1970- Forum Manager
- Messages : 485
Date d'inscription : 2015-05-14 -
Re: Quick Start with the wien2k code
![]() by Algerien1970 Fri 22 May - 18:46
by Algerien1970 Fri 22 May - 18:46
C/ Third stage : SCF calculation ( the calculation of the magnitudes : Enerrgy , Density and Others ...)
After the case has been set up, a link to ``run SCF'' is added, (``Run Programs $\rightarrow $ run SCF'' and you should invoke the self-consistency cycle (SCF). This runs the script run_lapw with the desired options.
The SCF cycle consists of the following steps:
LAPW0
(POTENTIAL) generates potential from density
LAPW1
(BANDS) calculates valence bands (eigenvalues and eigenvectors)
LAPW2
(RHO) computes valence densities from eigenvectors
LCORE
computes core states and densities
MIXER
mixes input and output densities
After selecting ``run SCF'' from the ``Execution'' menu, the SCF-window will open, and you can now specify additional parameters. For this example we select charge convergence to 0.0001: Specify ``charge'' to be used as convergence criterion, and select a value of 0.0001 (-cc 0.0001).
To run the SCF cycle, click on ``Run!''
Since this might take a long time for larger systems; you can specify the ``Execution type'' to be batch or submit (if your system is configured with a queuing system and w2web has been properly set up, see section 11.3).
While the calculation is running (as indicated by the status frame in the top right corner of the window), you can monitor several quantities (see section 3.9).
Once the calculation is finished (11 iterations), view case.dayfile for timing and errors and compare your results with the files in the provided example (TiC/case_scf).
For magnetic systems you would run a spin-polarized calculation with the script runsp_lapw. The program flow of such a calculation is described in section 4.5.2 and the script itself in section 5.1.3.
Algerien1970- Forum Manager
- Messages : 485
Date d'inscription : 2015-05-14 -
Re: Quick Start with the wien2k code
![]() by Algerien1970 Fri 22 May - 18:48
by Algerien1970 Fri 22 May - 18:48
Calculating Electronic Properties
Once the SCF cycle has converged one can calculate various properties like Density of States (DOS), band structure, Optical properties or X-ray spectra.
For the calculation of properties (which from now on will be called ``Tasks''). We strongly encourage the user to utilize the user interface, w2web. This user interface automatically supplies input file templates and shows how to calculate the named properties on a step by step basis.
Algerien1970- Forum Manager
- Messages : 485
Date d'inscription : 2015-05-14 -
Re: Quick Start with the wien2k code
![]() by Algerien1970 Sun 7 Jun - 18:35
by Algerien1970 Sun 7 Jun - 18:35
1 Electron density plots
Select ``El. Dens.'' from the ``Tasks'' menu and click on the buttons one by one (see figure 3.6):

The total charge density includes the Ti 3s and 3p states and the resulting density around Ti would be very large and dominated by these semicore states. To get a ``meaningful'' picture of the chemical bonding effects one must remove these states. Inspection of TiC.scf1 and TiC.scf2 should allow you to select an EMIN value to eliminate the Ti 3s and 3p semicore states.
Recalculate the valence density with EMIN=-1.0 to truncate Ti 3s and 3p (x lapw2). This is only possible, when you still have a valid TiC.vector file on a tetrahedral mesh.
Select a plane and plot the density in the (100) plane of TiC. When XCRYSDEN is installed, it will be offered automatically and provides a convenient way to specify a plane and create a colorful plot 3.7.
Select 2D-plot
Specify a resolution of 100 points (first line)
Select a plane by selecting 3 atoms and define these 3 atoms by clicking on them.
Choose rectangular parallelogram and enlarge the rectangular selection by 0.5 (for all 4 margins, then update the display)
calculate the density and produce a nice contour plot:
choose ``rainbow''-colors, activate all display-option buttens, and choose in ``Ranges'' a smaller ``highest rendered value''.
Finally, use smaller spheres (pipe+ball display model) and thinner bonds (Modify/Ball-Stick-ratio).
Alternatively, without XCRYSDEN, edit TiC.in5 and choose the offered template input file. To select the (100) plane for plotting specify the following input:
-1 -1 0 4 # origin of plot (x,y,z,denominator)
-1 3 0 4 # x-end of plot
3 -1 0 4 # y-end of plot
3 2 3 # x,y,z number of shells
100 100 # x, y plotting mesh, choose ratio similar to x,y length
RHO
ANG VAL NODEBUG
ORTHO
For a detailed description of input options consult section 8.6.3
Calculate electron density (x lapw5)
Plot output (using rhoplot), after the first preview select a range zmin=-0.5 to zmax=2

Compare the result with the electron density plotted in the (100) plane (see figure 3. . The program gnuplot (public domain) must be installed on your computer. For more advanced graphics use your favorite plotting package or specify other options in gnuplot (see rhoplot_lapw how gnuplot is called).
. The program gnuplot (public domain) must be installed on your computer. For more advanced graphics use your favorite plotting package or specify other options in gnuplot (see rhoplot_lapw how gnuplot is called).

Select ``El. Dens.'' from the ``Tasks'' menu and click on the buttons one by one (see figure 3.6):
Figure 3.6: Task ``Electron Density Plots''
The total charge density includes the Ti 3s and 3p states and the resulting density around Ti would be very large and dominated by these semicore states. To get a ``meaningful'' picture of the chemical bonding effects one must remove these states. Inspection of TiC.scf1 and TiC.scf2 should allow you to select an EMIN value to eliminate the Ti 3s and 3p semicore states.
Recalculate the valence density with EMIN=-1.0 to truncate Ti 3s and 3p (x lapw2). This is only possible, when you still have a valid TiC.vector file on a tetrahedral mesh.
Select a plane and plot the density in the (100) plane of TiC. When XCRYSDEN is installed, it will be offered automatically and provides a convenient way to specify a plane and create a colorful plot 3.7.
Select 2D-plot
Specify a resolution of 100 points (first line)
Select a plane by selecting 3 atoms and define these 3 atoms by clicking on them.
Choose rectangular parallelogram and enlarge the rectangular selection by 0.5 (for all 4 margins, then update the display)
calculate the density and produce a nice contour plot:
choose ``rainbow''-colors, activate all display-option buttens, and choose in ``Ranges'' a smaller ``highest rendered value''.
Finally, use smaller spheres (pipe+ball display model) and thinner bonds (Modify/Ball-Stick-ratio).
Alternatively, without XCRYSDEN, edit TiC.in5 and choose the offered template input file. To select the (100) plane for plotting specify the following input:
-1 -1 0 4 # origin of plot (x,y,z,denominator)
-1 3 0 4 # x-end of plot
3 -1 0 4 # y-end of plot
3 2 3 # x,y,z number of shells
100 100 # x, y plotting mesh, choose ratio similar to x,y length
RHO
ANG VAL NODEBUG
ORTHO
For a detailed description of input options consult section 8.6.3
Calculate electron density (x lapw5)
Plot output (using rhoplot), after the first preview select a range zmin=-0.5 to zmax=2
Figure 3.7: Electron density of TiC in (100) plane using Xcrysden
Compare the result with the electron density plotted in the (100) plane (see figure 3.
Algerien1970- Forum Manager
- Messages : 485
Date d'inscription : 2015-05-14 -
Re: Quick Start with the wien2k code
![]() by Algerien1970 Sun 7 Jun - 18:40
by Algerien1970 Sun 7 Jun - 18:40
2 Density of States (DOS)
Select ``Density of States (DOS)'' from the ``Tasks'' menu and click on the buttons one by one:
- Calculate partial charges (x lapw2 -qtl). (This is only possible, when you still have a valid TiC.vector file on a tetrahedral mesh.)
- Edit TiC.int, choose the offered template input file and edit it to select: total DOS, Ti-d, Ti-d
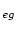 , Ti-d
, Ti-d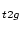 , C-s and C-p-like DOS.
, C-s and C-p-like DOS.
TiC
-0.50 0.00200 1.500 0.003 EMIN, DE, EMAX, Gauss-broadening
6 NUMBER OF DOS-CASES
0 1 tot (atom,case,description)
1 4 Ti d
1 5 Ti eg
1 6 Ti t2g
2 2 C s
2 3 C p
For a detailed description of input options consult section 8.1.3 - Calculate DOS (x tetra).
- Preview output using ``dosplot''
If you want to use the supplied plotting interface dosplot2 to preview the results, the program gnuplot (public domain) must be installed on your computer.
The calculated DOS can be compared with figures 3.9 and 3.10. Together with the electron density the partial DOS allows you to analyse the chemical bonding (covalency between
 and
and  , non-bonding
, non-bonding  , charge transfer estimates,....)
, charge transfer estimates,....) 
Figure 3.9: Density of states of TiC

Figure 3.10: Density of states of TiC
Algerien1970- Forum Manager
- Messages : 485
Date d'inscription : 2015-05-14 -
Re: Quick Start with the wien2k code
![]() by Algerien1970 Sun 7 Jun - 18:42
by Algerien1970 Sun 7 Jun - 18:42
3 Bandstructure
Select ``Bandstructure'' from the ``Tasks'' menu and click on the buttons one by one:- Create the file TiC.klist_band from the template in $WIENROOT/SRC_templates/fcc.klist. (To calculate a bandstructure a special k-mesh along high symmetry directions is necessary. For a few crystal structures template files are supplied in the SRC-directory, you can also use XCRYSDEN (save it as xcrysden.klist) to generate a k-mesh or type in your own mesh.
- Calculate Eigenvalues using the ``-band'' switch (which changes lapw1.def such that the k-mesh is read from TiC.klist_band and not from TiC.klist)
Note: When you want to calculate DOS, charge densities or spectra after this bandstructure, you must first recalculate the TiC.vector file using the ``tetrahedral'' k-mesh, because the k-mesh for the band structure plots is not suitable for calculations of such properties. - Edit TiC.insp: insert the correct Fermi energy (which can be found in the saved scf-file) and specify plotting parameters. For comparison with figure 3.12 select an energy-range from -13 to 8 eV.
- Calculate Bandstructure (x spaghetti).
- Preview Bandstructure (needs ghostscript installed).
If you want to preview the bandstructure, the program ghostview (public domain) must be installed on your computer. You can compare your calculated bandstructure with figure 3.12.

Figure 3.12: Bandstructure of TiC
Algerien1970- Forum Manager
- Messages : 485
Date d'inscription : 2015-05-14 -
» Exercises_08 : wien2k
» The version of 2012 of wien2k code
» The version of 2014 of wien2k code
» The band gap problem: the accuracy of the Wien2k code confronted
» WIEN2k-Textbooks:
» The version of 2012 of wien2k code
» The version of 2014 of wien2k code
» The band gap problem: the accuracy of the Wien2k code confronted
» WIEN2k-Textbooks:
Calculations of Solid Properties Using Wien2k Package :: WIEN2K ENVIRONMENT :: WIEN2K DOCUMENTATIONS
Page 1 of 1
Permissions in this forum:
You cannot reply to topics in this forum|
|
|